Research Themes
Research in the Bik Lab is intensely interdisciplinary, using high-throughput sequencing and diverse –Omics approaches to explore broad patterns in marine microbes (biodiversity and phylogeography, functional roles for microbial taxa, and the relationship between species and environmental parameters), with an emphasis on microbial eukaryotes in marine sediment habitats. Our long-term research interests lie at the interface between biology and computer science, using biological questions and evolutionary hypotheses to drive the development and refinement of –Omic approaches focused on marine microbes.
Lab research themes span three key areas:

Nematode feeding groups (based on morphology) do not correlate with microbiome distinctness (Schuelke et al. 2018)
1. Merging microbiome sequencing and biogeochemical measurements to elucidate trophic transfer in marine sediments. Nematode worms are populous members of sediment habitats worldwide (from coastal estuaries to deep-sea methane seeps), but little is known regarding their interactions with sediment geochemistry and bacteria and archaeal members of the community. Recent work from the lab (Schuelke et al. 2018, Molecular Ecology) suggests that nematode worms may be feeding on prokaryotic taxa that play important roles in global biogeochemical cycles (methane, sulfur, and nitrogen). Microbiome profiles (16S rRNA amplicons) obtained from single worms indicate the presence of diverse taxa in nematode gut tracts, including methylotrophic and methanotrophic bacteria, ammonia-oxidizing bacteria and archaea, sulfate-reducing bacteria, and Beggiatoa species (the dominant mat-forming bacteria at methane seeps). These microbiome patterns were geographically widespread, and were observed in nematodes isolated from Arctic background sediments to Gulf of Mexico methane seeps. Furthermore, nematodes appear to be selectively ingesting a subset of prokaryotic taxa present in marine sediments: nematode gut contents clustered separately and distinctly from overall bacterial/archaeal assemblages in bulk sediments. Our current hypothesis is that marine nematodes may be selectively feeding on bacterial/archaeal taxa based on chemosensory information (targeting prokaryotic taxa that produce certain cellular lipids or metabolites).

The Phinch 2.0 desktop app - http://phinch.org
2. Advancing the pace of environmental microbiome studies via new computational frameworks and data visualization tools. The vast majority of -Omics software development is driven by biomedical studies of bacteria/archaea, with many common bioinformatics workflows originating from the human microbiome and infectious disease fields. A second lab focus aims to address this research gap, by making rapid advances in the bioinformatics toolkits available for environmental microbiome studies (e.g. focused on non-model organisms and natural environments). Software development is inherently complementary to our hypothesis-driven research objectives. A primary focus is the development of computational pipelines that promote scientific efficiency (rapid data exploration and quick generation of results and figures, e.g. the Phinch data visualization framework) and specifically account for the genome structure of microbial eukaryotes (accounting for intragenomic variation in multicopy rRNA genes, and finding workarounds for sparse genome databases).
~300 sampling locations being analyzed as part of the 5-year Southern California Bight Survey (collaboration with SCCWRP)
3. Population biology, genome evolution, and biomonitoring applications of marine microbiome datasets. Advances in environmental –Omics approaches (metabarcoding, metagenomics, and metatranscriptomics) now enable the assessment of population-level patterns in deep sequencing datasets. For example, the increasing use of multi-gene metabarcoding studies (e.g. incorporating both nuclear and organelle markers) and incorporation of phylogenetic placement approaches are now providing insights on genetic connectivity and evolutionary signatures of whole microbial communities. These approaches have direct relevance to environmental biomonitoring applications, whereby microbiome structure and diversity can provide rapid information on ecosystem health. As a third research focus, the Bik Lab is using multi –Omic sequencing approaches (primarily metagenomics and multi-marker metabarcoding) to assess environmental drivers of microbial community structure in marine sediments. Two large ongoing projects focus on Arctic sediments in the Beaufort and Chuckchi Seas, and offshore and intertidal sediments in the Southern California Bight. Both projects involve hundreds of samples (>200 per project) and a detailed suite of environmental metadata collected from each sample site. In Arctic sediments, we are assessing how depth, local hydrography, sediment grain size, and the quality/quantity of organic matter impacts the biodiversity and distribution of microbial taxa. In the California Bight, the primary focus is to determine microbial bioindicator species indicative of heavily polluted sites such as the Port of Los Angeles (sediments with high levels of legacy pesticides such as PCBs and DDT), as well as assess the importance of emerging pollutants such as microplastics in determining microbial community structure and function. The biodiversity of marine sediments is poorly characterized in general (in contrast to water column microbiomes which have benefited from large global initiatives such as TARA oceans and the Global Ocean Sampling expedition), and thus our Arctic and California sediment datasets represent important baseline surveys of microbial biodiversity. In addition, metagenome sequencing will facilitate the assembly and binning of Metagenome Assembled Genomes (MAGs) isolated from marine sediments, and these genome sequences will be used to conduct deeper studies of microbial genome evolution and adaptation to life in polar regions and heavily polluted sediments.
Active Projects
LOFRESH: Understanding the ecological relevance of eDNA in freshwater lotic ecosystems
LOFRESH is an international collaborative initiative funded by a UK Natural Environment Research Council (NERC) Highlight Topic grant to improve our ability to utilize environmental DNA (eDNA) for tracking the presence and abundance of species in and around freshwater habitats. Environmental DNA refers to shed cells or extracellular DNA from organisms as they pass through an environment (e.g. water, soil and air), or die and decay. By doing so, animals and plants leave traces of their DNA in the environment that can be detected using a number of molecular genetic approaches.
We expect that our findings will provide valuable insights for the fields of freshwater ecology, biomonitoring and environmental assessment. Launched in March 2016, LOFRESH aims to understand the dynamics between living communities and lotic (i.e. riverine) eDNA in relation to spatial and environmental variation.
The LOFRESH project is led by Prof. Simon Creer at Bangor University, and also includes a multi-institutional team of UK collaborators from the The Centre for Ecology and Hydrology (CEH), Cardiff University, and the University of Birmingham.
References:
Seymour M, Durance I, Cosby BJ, Ransom-Jones E, Deiner K, Ormerod SJ, Colbourne JK, Wilgar G, Carvalho GR, de Bruyn M, Edwards F, Emmett BA, Bik HM, Creer S (2018) Acidity promotes degradation of multi-species environmental DNA in lotic mesocosms, Communications Biology, 1(1):4. (editors’ picks for one year anniversary collection; open access publication)
Genomic responses to the Deepwater Horizon event and development of high-throughput biological assays for oil spills
This is a collaborative project led by Kelley Thomas at the University of New Hampshire; the team of Co-PIs includes the Bik Lab, Paul Montagna the Harte Research Institute for Gulf of Mexico Studies (Texas A&M), and Jon Norenburg at the Smithsonian National Museum of Natural History. Our grant is funded through the Gulf of Mexico Research Initiative, and a detailed overview of the award can be found on the GOMRI website.
The overall aim of this project is to improve the response, mitigation, detection, characterization and remediation associated with oil spills and accompanying release of gas.
The benthic environment in the Gulf of Mexico (GoM) is biologically hyper-diverse, performing critical ecosystem functions that have consequences for the ecology of the entire GoM region. Benthic communities are strongly impacted by oil spills, which render them a valuable tool for assaying and monitoring the impacts of contamination. However, detailed and extensive characterization of these communities has been impractical for due to the tedious and time-consuming nature of the taxonomic efforts required to accurately describe small benthic fauna. Our project leverages high-throughput sequencing technologies that now enable rapid, accurate, and cheap assays of community biodiversity. To achieve these goals, our GOMRI project team brings together the interdisciplinary expertise in marine biology, taxonomy, genomics and bioinformatics necessary for the development of a meaningful and robust technology. Project goals include three main objectives:
Use targeted sequencing of individual benthic eukaryotes to generate a representative sample of diverse genomes from which to select an expanded set of nuclear and mitochondrial loci for targeted mining of shotgun metagenomic data.
Assess eukaryotic community structure across space and time via high-throughput sequencing of environmental metagenomes using a new and expanded array of nuclear and mitochondrial marker genes.
Establish Standard Operating Procedures (SOPs) and reproducible bioinformatic workflows for environmental monitoring of oil spills. This will include establishing a database for integration of taxonomic and molecular datasets, and dissemination of tools and educational resources.
Training the next generation of environmental biologists with interdisciplinary tools, via two yearly workshops focused on high-throughput sequencing workflows. These workshops will expose students to the full spectrum of this technology from sample preparation, through taxonomy, to metagenomics and bioinformatics. These workshops are also opportunities to attract underrepresented groups and to link the research team with GoM stakeholders. All workshops will be held at the Harte Research Institute for Gulf of Mexico Studies at Texas A&M University, Corpus Christi.
References and Resources:
2nd Benthic Invertebrates, Metagenomics, and Bioinformatics Workshop in Corpus Christi, Tx (January 15-19, 2018) - 2018 workshop agenda and tutorials on GitHub
Benthic Invertebrates, Metagenomics, and Bioinformatics Workshop in Corpus Christi, Tx (January 9-13, 2017) - 2017 workshop agenda and tutorials on GitHub
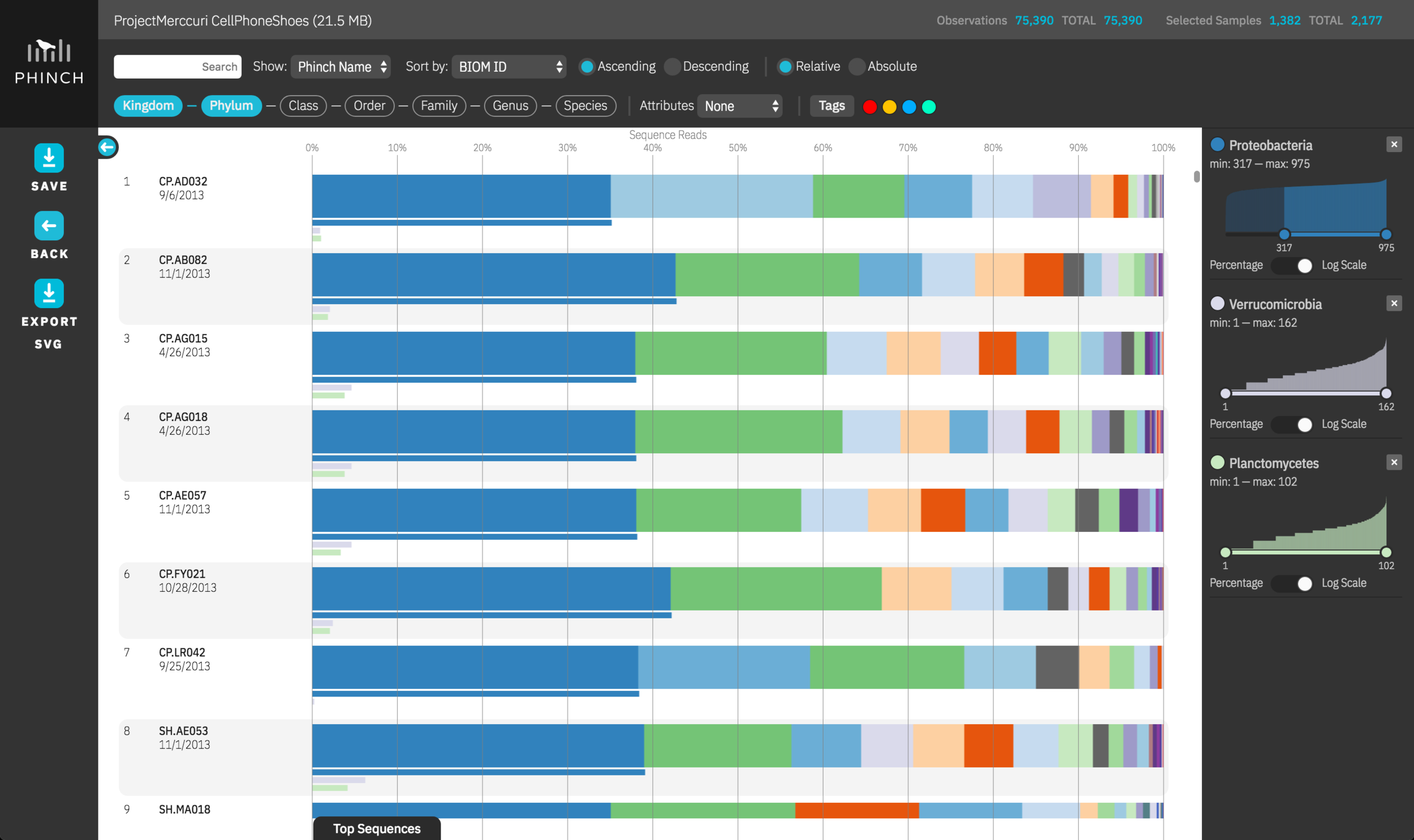
Phinch: A research-driven, exploratory data visualization framework for environmental -Omics data
Phinch is an open-source data visualization framework aimed at promoting novel explorations of large biological datasets (microbiomes, metagenomes, etc.). This project represents an interdisciplinary collaboration between Pitch Interactive (a data visualization studio in Oakland, CA) and the Bik Lab at UC Riverside (grant PI: Holly Bik), and has been funded via two grants from the Alfred P. Sloan Foundation.
The new Phinch 2.0 software has been relaunched as a standalone desktop application, compatible with Mac OS X, Windows, and Linux operating systems. New features include support for HDF5 and JSON BIOM files, retooled visualizations, improved image export and sharing, and the ability to work offline. The open-source codebase, software documentation, and bug/issue tracking can be found on the Phinch 2.0 app GitHub repository.
References:
Bik HM, Pitch Interactive (2014) Phinch: An interactive, exploratory data visualization framework for –Omic datasets, bioRxiv, doi:10.1101/009944 (preprint)

Genomic baseline surveys of Arctic marine sediments
This project is a collaboration with Sarah Hardy at the University of Alaska, Fairbanks, funded through an award from the North Pacific Research Board. This research focuses on a set of >100 marine sediment samples collected from two ocean regions (the Beaufort and Chuckchi Seas) off the north slope of Alaska. We are collecting and comparing three types of independent datasets: 1) Morphological taxonomy data from nematode communities, 2) Environmental marker gene amplicons (16S and 18S rRNA) to broadly examine bacteria, archaea, and microbial eukaryote communities, and 3) Shotgun metagenomic data as a “PCR-free” approach for describing microbial community assemblages. Our goal is to compare biological insights gained from traditional taxonomy vs. high-throughput genomic datasets, and to assess meiofaunal community diversity and phylogeographic patterns in understudied Arctic regions.
References:
Schuelke T, Pereira TJ, Hardy SM, Bik HM (2018) Nematode-associated microbial taxa do not correlate with host phylogeny, geographic region or feeding morphology in marine sediment habitats, Molecular Ecology, 27(8): 1930-1951. (invited submission for “Host-Associated Microbiome” special issue; PDF Download)

NSF Research Coordination Network EukHiTS
RCN EukHiTS (Eukaryotic biodiversity research using High-Throughput Sequencing) is a collaborative project between the Bik Lab at UC Riverside and Kelley Thomas’s lab at University of New Hampshire. EukHiTS is funded through a Research Coordination Network award from the National Science Foundation (DBI-1262480); the goal of this project is to catalyze formation of an international scientific network focused on -Omic investigations of microbial eukaryotes and promote cross-disciplinary interactions.
Microscopic eukaryote species (organisms <1mm, such as nematodes, fungi, protists, etc.) are abundant and ubiquitous, yet invisible to the naked eye, in every ecosystem on earth. The biodiversity and geographic distributions for most of these species are largely unknown, and represent one of the major knowledge gaps in biology. High-throughput DNA sequencing technologies now allow for deep examination of virtually all microscopic organisms present in an environmental sample. For microbial eukaryote taxa, en masse biodiversity assessment using traditional loci (rRNA genes) can be conducted at a fraction of the time and cost required for traditional (morphological) approaches. Despite this promise, current bottlenecks include the lack of useful distributed tools for analysis and common data standards to allow global comparisons across individual studies as well as missing links between molecules and morphology. RCN EukHiTS is focusing on developing community capabilities for computational approaches focused on eukaryotic taxa and the infrastructure, both cyber and human, needed for effective interpretation of large high-throughput datasets. The steering committee of RCN EukHiTS includes expertise from computational biology, functional genomics, computer science, taxonomy, ecology, database resource management, and representatives of end user communities to ensure that all aspects of the community are well-represented.
This NSF RCN builds on two previous community meetings organized by PIs Holly Bik and Kelley Thomas: a 2014 SMBE Satellite Meeting on Eukaryotic -Omics and a 2011 NESCent Catalysis Meeting on “High-Throughput Biodiversity Assessment using Eukaryotic Metagenetics”.
References:
Deiner K, Bik HM, Mächler E, Seymour M, Lacoursière-Roussel A, Altermatt F, Creer S, Bista I, Lodge DM, de Vere N, Pfrender ME, Bernatchez L (2017) Environmental DNA metabarcoding: transforming how we survey animal and plant communities, Molecular Ecology, 26(21): 5872-5895. (invited review; open access publication)
Creer S, Deiner K, Frey S, Porazinska D, Taberlet P, Thomas WKT, Potter C, Bik HM (2016) The ecologist’s field guide to sequence-based identification of biodiversity, Methods in Ecology and Evolution, 7(9): 1008-1018. (invited review; open access publication)
Bik HM, Porazinska DL, Creer S, Caporaso JG, Knight R, Thomas WK (2012) Sequencing our way towards understanding global eukaryotic biodiversity. Trends in Ecology and Evolution, 27(4):233-243. (PDF download)
Completed Projects

PressForward (External Collaboration)
PressForward is a software tool that enables curation and sharing of content from around the internet. This free wordpress plugin works to collect information via an RSS feed reeder and browser bookmarklet, amalgamating, republishing and sharing content in one central location (typically via blog feed). The PressForward project is being led Stephanie Westcott at the Roy Rosenzweig Center for History and New Media at George Mason University.
As a PressForward Pilot Partner, we have guided the installation of this software tool on the microBEnet and Deep-Sea Biology Society websites, and helped build a team of community editors to review and disseminate content on a daily basis. Our goal as a Pilot Partner is to reduce the manual effort required for website administration (minimizing the administrative burden for scientists maintaining project websites), and to aggregate and share content in a way that is not currently possible with the default Wordpress backend.

microBEnet (External Collaboration)
microBEnet (the Microbiology of the Built Environment Network - http://www.microbe.net) is an online portal for resources related to the microbiology of the built environment. This project is led and maintained by Jonathan Eisen’s lab at the University of California, Davis, and funded by a grant from the Alfred P. Sloan Foundation. The microBEnet project has historically focused on three main categories of tasks, including 1) organizing meetings and workshops, 2) leveraging social media to facilitate communication and collaboration, 3) curating and creating online resources to facilitate work in the Built Environment and to build a culture of openness and sharing. PI Holly Bik has been involved with curating online resources and social media tools for microBEnet, as well as providing bioinformatics oversight and design advice for microBEnet’s citizen science and undergraduate research projects.
References:
Bik HM, Alexiev A, Aulakh S, Bharadwaj L, Flanagan J, Haggerty M, Hird S, Jospin G, Lang JM, Sauder L, Neufeld J, Shaver A, Sethi A, Eisen JA, Coil D. Microbial community succession and nutrient cycling responses following perturbations of experimental saltwater aquaria, mSphere, 4 (1) e00043-19. (open access publication)
Bik HM, Coil D, Eisen JA (2014) microBEnet: Lessons learned from building an interdisciplinary scientific community in the online sphere, PLoS Biology, 12(6): e1001884. (open access publication)
PhyloSift (External Collaboration)
PhyloSift is a software pipeline for phylogenetic analysis of genomes and metagenomes. Using any biological sequence as input data (nucleotide or amino acid), PhyloSift uses a reference database of profile HMMs to identify candidate sequences matching phylogenetically-informative marker genes. Candidate sequences identified from input data are then subjected to phylogenetic placement approaches, where short reads are inserted into reference phylogenies and given taxonomic assignments based on this tree placement. PI Holly Bik was actively involved in development and documentation of the PhyloSift pipeline, and we continue to use this software for ongoing metagenomics data analysis. Software download and extensive documentation are available on the main PhyloSift website, and the code is maintained as an open source repository on Github.
References:
Darling A, Jospin G, Lowe E, Matsen FA, Bik HM, Eisen JA (2014) PhyloSift: phylogenetic analysis of genomes and metagenomes, PeerJ, 2:e243. (open access publication)